Hipercolesterolemia familiar
Descripción general
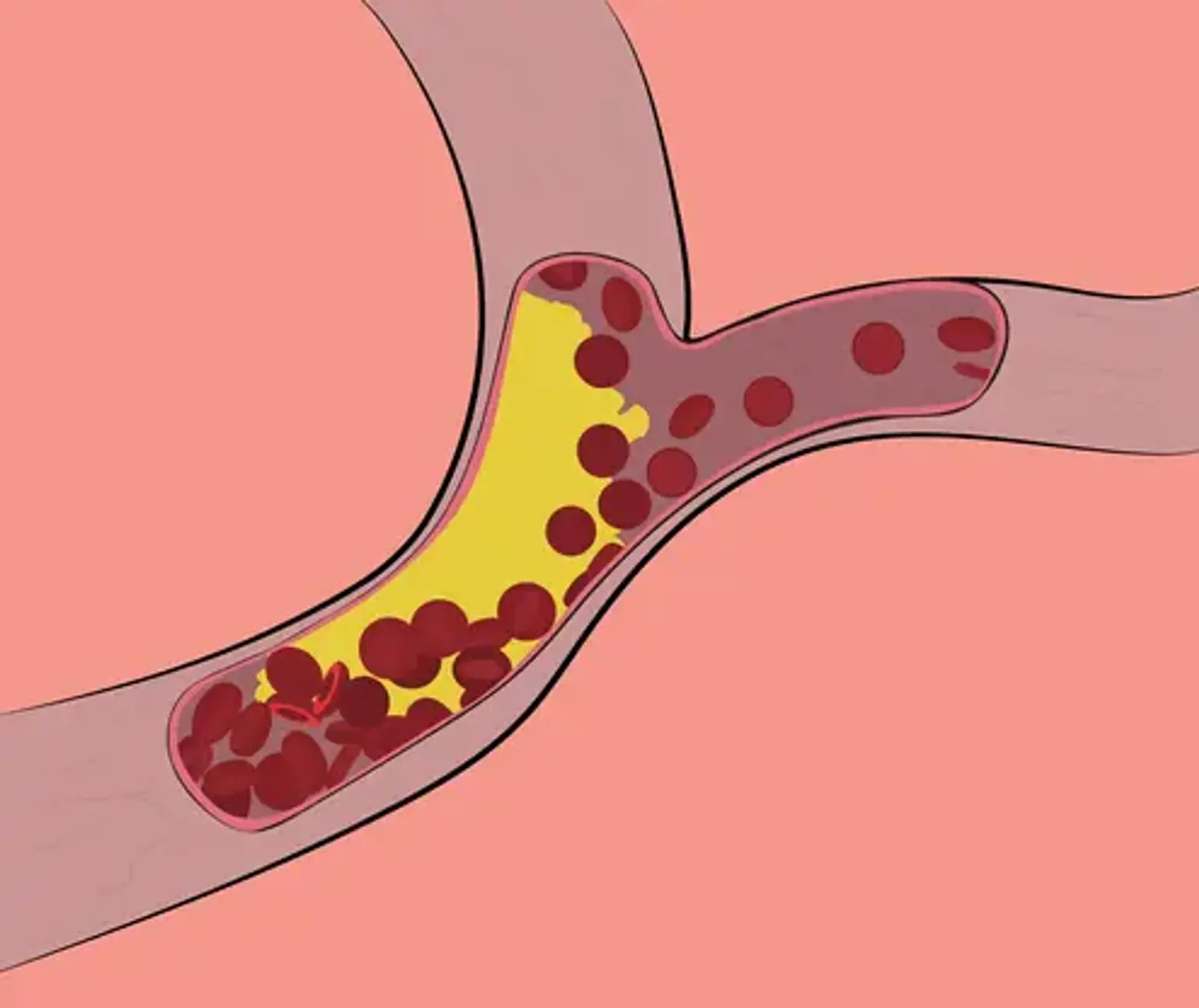
La hipercolesterolemia familiar es un conjunto de anomalías genéticas hereditarias que provocan un aumento significativo del nivel de colesterol en sangre. Aunque la aterosclerosis por HF se desarrolla principalmente en la edad adulta, puede comenzar ya en la primera década de la vida. El hecho de que el tratamiento temprano de los factores de riesgo pueda revertir las alteraciones ateroscleróticas en el sistema vascular enfatiza la necesidad de detectar y tratar a los niños con esta enfermedad lo antes posible.
¿Qué es la hipercolesterolemia familiar?
La hipercolesterolemia familiar (FH) es una enfermedad genética que afecta la forma en que el cuerpo recicla el colesterol LDL (malo). Como resultado, los niveles de LDL en sangre siguen siendo extremadamente altos; en casos graves, los niveles pueden exceder los 190 miligramos por decilitro (mg/dL)
Las personas que tienen HF nacen con colesterol LDL elevado. Los niveles de colesterol de todas las personas tienden a aumentar a medida que envejecen. Sin embargo, los pacientes con HF tienen niveles de LDL que comienzan siendo altos y aumentan con el tiempo.
La última década ha visto un aumento en los estudios sobre el origen genético y el tratamiento de esta enfermedad. Sin embargo, debido a la falta de comprensión entre los pediatras y el público en general, la HF con frecuencia se diagnostica erróneamente hasta que se han establecido los efectos irreversibles de la aterosclerosis.
Esto, al igual que las anomalías del colesterol no heredadas, se suma a las placas ateroscleróticas, lo que aumenta significativamente el riesgo de enfermedad coronaria . Las personas con HF tienen 20 veces más probabilidades de desarrollar una enfermedad cardíaca si no reciben tratamiento.
Los hombres con HF desarrollan enfermedad coronaria entre 10 y 20 años antes. Antes de los 50 años, la mitad de los hombres con HF no tratada sufrirán un ataque cardíaco o angina . Algunos tendrán poco más de veinte años. La enfermedad coronaria puede ocurrir hasta 20 o 30 años antes en las mujeres. Aproximadamente el 30% de las mujeres que no reciben tratamiento sufrirán un ataque cardíaco antes de los 60 años.
Estos mayores riesgos no están relacionados con otros factores de riesgo, que podrían exacerbar la situación. La buena noticia es que la FH se puede controlar con una combinación de modificaciones en el estilo de vida y medicamentos.
¿Qué tan frecuente es la Hipercolesterolemia Familiar (HF)?
Según investigaciones recientes, la HF es frecuente en 1 de cada 250 personas, lo que la convierte en una de las enfermedades hereditarias más comunes. Sin embargo, dado su importante riesgo, la mayoría de las personas reciben un diagnóstico erróneo y un tratamiento insuficiente.
Se cree que 1 de cada 160.000 a 1.250.000 personas padece hipercolesteremia familiar (HF). Es más probable que la HF se desarrolle en países con una alta frecuencia de HF, particularmente aquellos con una alta tasa de consanguinidad (matrimonio entre primos).
Trastornos relacionados
La HF se puede diagnosticar utilizando cualquiera de los criterios de diagnóstico estandarizados establecidos que se enumeran en la sección "Diagnóstico". Cuando se sospecha un diagnóstico de HF pero no se puede confirmar completamente según la evidencia disponible, las pruebas genéticas con frecuencia pueden ayudar. En 2019, un panel internacional de expertos emitió un documento de consenso a favor del valor de las pruebas genéticas de HF, así como una declaración que indica que las pruebas genéticas deben administrarse como estándar de atención.
Cuando las pruebas genéticas son negativas, los límites tecnológicos y la sensibilidad de las pruebas genéticas de HF no pueden descartar definitivamente la HF, pero pueden promover el examen de razones alternativas para los síntomas de presentación. Hay otras enfermedades que tienen resultados de laboratorio o aspectos de antecedentes familiares comparables a los de la HF. Estos son algunos ejemplos:
- La obesidad , la diabetes mellitus , el hipotiroidismo , los medicamentos como los esteroides o la enfermedad renal pueden causar hipercolesterolemia. El patrón de herencia no es mendeliano.
- La hipercolesterolemia poligénica es causada por una serie de variaciones genéticas de impacto menor en una variedad de genes que metabolizan el colesterol. Los factores de riesgo como la diabetes y la obesidad pueden agravar la hipercolesterolemia poligénica. Un individuo puede tener antecedentes familiares de hiperlipidemia y enfermedad de las arterias coronarias, incluso si la aparición de CAD ocurre más tarde en la vida. La herencia sigue un patrón no mendeliano y los miembros de la familia frecuentemente aparecen con expresión variable de niveles de LDL-C o riesgos cardiovasculares.
- La lipoproteína a (Lp) es una partícula similar al colesterol que, cuando aumenta (>30 mg/dL), aumenta el riesgo de enfermedad arterial coronaria (EAC) temprana. Los niveles de lipoproteínas están influenciados principalmente por la genética y se heredan de forma autosómica codominante. Las personas con niveles muy elevados de lipoproteínas pueden tener antecedentes familiares similares a los de alguien con HF, incluso si sus niveles de colesterol LDL son normales. Las personas que tienen tanto FH como un exceso de lipoproteínas tienen un mayor riesgo de desarrollar enfermedad arterial coronaria temprana.
Fisiopatología de la HF
La expresión fenotípica del metabolismo defectuoso de las lipoproteínas inducida por una serie de trastornos genéticos se conoce como hipercolesterolemia familiar. Tras el importante hallazgo de Brown y Goldstein de que las mutaciones en el receptor de lipoproteínas de baja densidad (LDLR) eran la causa de la HF monogenética, se han identificado aproximadamente 1.500 mutaciones en este gen, lo que representa más del 80% de los casos de HF monogenética.
La lipoproteína de baja densidad (LDL) transporta la mayor parte del colesterol plasmático mediante su unión a la membrana celular LDLR mediante dos ligandos en la LDL, la apolipoproteína B-100 (apoB-100) y la apoE. La combinación LDL/LDLR penetra en las células, luego se libera LDL y el LDLR se recicla a la membrana celular.
El hígado secreta la proproteína PCSK9, que se une extracelularmente al LDLR e impide su reciclaje a la membrana celular. Como resultado, PCSK9 disminuye el LDLR mientras aumenta el LDL sérico. El efecto final de estas tres mutaciones genéticas principales es un mal funcionamiento de la unión de los receptores de LDL al colesterol LDL, lo que resulta en una disminución en la absorción y destrucción del colesterol LDL en el hígado y un aumento en los niveles de LDL en sangre. Clínicamente, es imposible identificar la mutación genética subyacente en la hipercolesterolemia familiar.
Síntomas de hipercolesterolemia familiar
Se requiere una historia familiar completa para la evaluación y el diagnóstico de la hipercolesterolemia familiar. Los familiares de primer grado con antecedentes familiares de enfermedad arterial coronaria temprana o colesterol alto (menores de 60 años en mujeres y 55 años en hombres) son señales de alerta. También debería plantear la posibilidad de hipercolesterolemia familiar en individuos jóvenes que tienen familiares de segundo grado que han tenido episodios coronarios tempranos.
Tipos de hipercolesterolemia familiar
-
Hipercolesterolemia familiar homocigótica
Estos individuos tienen síntomas de cardiopatía isquémica, enfermedad vascular periférica, enfermedad cerebrovascular o estenosis aórtica. Puede haber tendinitis o artralgia, así como antecedentes de lesiones cutáneas extrañas. La supervivencia después de los 30 años es difícil a menos que se utilicen procedimientos no convencionales.
-
Hipercolesterolemia familiar heterocigótica
Desde la infancia, estos individuos han tenido una hipercolesterolemia significativa. Los síntomas de la cardiopatía isquémica están muy extendidos, especialmente cuando están presentes factores de riesgo cardiovascular adicionales. Puede haber tendinitis de Aquiles recurrente o síntomas artríticos.
Examen físico
Por lo general, las pruebas físicas anormales están asociadas con depósitos de colesterol en los ojos o la piel. En la hipercolesterolemia familiar homocigótica, puede ocurrir a una edad temprana. Los xantomas tendinosos, que aparecen como engrosamiento de los tendones debido al colesterol acumulado entre los macrófagos en el tejido conectivo, son patognomónicos de la hipercolesterolemia familiar. El tendón de Aquiles y los tendones extensores de los dedos son los más comúnmente afectados, pero también son frecuentes los tendones rotuliano y del tríceps.
Los xantomas tendinosos afectan a menos de la mitad de los pacientes con HF. El arco senilis corneal son depósitos de colesterol que rodean el borde corneal. Es más específico en pacientes menores de 45 años. El depósito de colesterol también puede tomar la forma de xantomas tuberosos de color amarillo anaranjado (en las palmas, los codos o las rodillas) o xantelasma (en los párpados), que son más específicos de la HF en los pacientes menores de 45 años. personas de 20 a 25 años.
Diagnóstico de hipercolesterolemia familiar
Los niños no tratados con niveles de LDL-C superiores a 160 mg/dL, o con niveles de LDL-C superiores a 130 mg/dL con antecedentes familiares positivos de HF o enfermedad cardíaca prematura, deben ser evaluados para detectar HF. Un C-LDL superior a 190 mg/dL, antecedentes personales y/o familiares de EAC temprana, indicaciones físicas como las indicadas en "Síntomas" o un familiar que se sabe que tiene HF deben hacer sospechar de HF en adultos no tratados.
Se descubre una variación de FH aproximadamente entre el 60 y el 80 % de las veces en personas sospechosas de tener FH según criterios clínicos. Aunque las pruebas de ADN se consideran el "estándar de oro", no siempre son necesarias ni posibles. Cuando no está claro si un individuo está afectado o no, se deben explorar las pruebas de ADN, y son especialmente útiles para realizar pruebas a miembros de la familia. Investigaciones recientes también sugieren que los riesgos individuales de CAD difieren según el gen afectado y el tipo de mutación del ADN.
Los individuos con un C-LDL >190 mg/dL y una variación patogénica de FH tienen un riesgo relativo diez veces mayor de enfermedad coronaria (en comparación con la población general), mientras que aquellos con un C-LDL >190 mg/dL pero sin mutación patogénica de FH tienen un riesgo relativo tres veces mayor de CAD (en comparación con la población general). Las personas que dan positivo en las pruebas genéticas pueden tener un mayor riesgo de enfermedad coronaria que aquellas que dan negativo en las pruebas genéticas en cualquier nivel de LDL-C determinado.
Una vez que a una persona se le ha diagnosticado HF, se recomienda un proceso conocido como "detección en cascada", "prueba en cascada" o "detección familiar" (pruebas de familiares cercanos) para identificar a las personas con HF antes de que aparezcan los síntomas, lo que permite una evaluación temprana e intensiva. comenzar el tratamiento y evitar la enfermedad y la muerte. Si se encuentra una variación patogénica, el riesgo en los parientes de primer grado del paciente (padres, hermanos, hijos) y, si es necesario, en los parientes más lejanos se puede determinar mediante pruebas de ADN mediante el seguimiento del gen modificado a través de la familia.
Si no se realizan pruebas de ADN, se puede utilizar una variación de la detección en cascada basada en el colesterol. Se ha demostrado que la detección en cascada, mediante cualquier método, es útil para identificar personas con HF que no estaban siendo tratadas adecuadamente. Un asesor genético puede ayudar a una familia con este procedimiento.
Pruebas clínicas y diagnóstico
Evaluaciones tras el diagnóstico inicial.
Las siguientes evaluaciones están indicadas en adultos y niños para determinar la magnitud de la enfermedad y los requerimientos de un individuo diagnosticado con HF:
- Niveles de lípidos antes de la terapia.
- Los niveles de lipoproteína (a) deben medirse siempre que sea posible, ya que la lipoproteína (a) es un factor de riesgo adicional para la enfermedad coronaria.
- Exclusión de condiciones concomitantes que puedan afectar las lecturas de lípidos ( enfermedad renal , hipotiroidismo no tratado, infarto agudo de miocardio, infección).
- El colesterol total (CT), el colesterol unido a lipoproteínas de baja densidad (C-LDL), el colesterol unido a lipoproteínas de alta densidad (C-HDL) y los triglicéridos son todos parte del panel de lípidos.
- Consulta con especialista en lípidos o médico con conocimientos en FH Genética médica
Algunas pautas proponen técnicas de imagen no invasivas (p. ej., evaluación del espesor de la íntima-media carotídea) en niños para ayudar a informar las opciones de tratamiento.
Gestión
El tratamiento de la FH se centra en reducir los niveles de LDL-C para reducir el riesgo de enfermedad cardíaca aterosclerótica.
Adultos con HF
-
Intervención en el estilo de vida
Los ajustes dietéticos , como limitar las grasas saturadas y eliminar las grasas trans, tienen una influencia importante en los niveles de colesterol. También son beneficiosos reducir el colesterol dietético y aumentar la fibra soluble. Las verduras, frutas y cereales enteros, frutos secos y legumbres deben ser los pilares de la dieta. Las mejores fuentes de proteína animal incluyen mariscos, pollo magro y productos lácteos bajos en grasa. La reducción de peso y el ejercicio aeróbico tienen muy poco impacto en los niveles de colesterol, pero pueden ayudar a disminuir la presión arterial y los niveles de azúcar en sangre y, por tanto, el riesgo de enfermedad cardiovascular.
-
Medicamentos para bajar el colesterol
Los adultos deben comenzar la terapia lo antes posible después del diagnóstico. Casi todos necesitarán medicación para reducir el colesterol. Un diagnóstico positivo de HF debería impulsar una terapia más intensiva que la que normalmente se administraría a un paciente con colesterol alto "tipo jardín". Algunas pautas recomiendan que los niveles de colesterol no tratados se reduzcan al menos en un 50%; otros recomiendan que aquellos sin un incidente previo de ECV intenten consumir menos de 100 mg/dL. Cuando están presentes otros factores de riesgo como diabetes o aterosclerosis, los objetivos de C-LDL son más severos (normalmente 70 mg/dl). Si estos objetivos no se pueden alcanzar en el contexto de atención primaria, los pacientes con HF deben ser derivados a un lipidólogo.
La farmacoterapia debe comenzar con estatinas, seguida de la inclusión de medicamentos adicionales si no se obtiene el nivel deseado de LDL-C con estatinas y ajustes en el estilo de vida. Se debe preferir una de las estatinas de mayor potencia (atorvastatina o rosuvastatina) administrada en la dosis máxima.
El daño muscular (rabdomiólisis) es el efecto secundario más común de las estatinas, pero ocurre sólo en aproximadamente 1 de cada 10.000 personas que las toman. El daño al hígado no ocurre con mayor incidencia en los usuarios de estatinas que en los que no las toman. La mialgia (malestar muscular) es un efecto secundario moderadamente frecuente que afecta al 10-15% de las personas. La mialgia menor, con o sin elevaciones leves de la creatina quinasa (menos de 5 veces el límite superior normal), no siempre es motivo para dejar de usar estatinas u otros medicamentos para reducir el colesterol.
Es posible que se requieran otros medicamentos, como ezetimiba (Zetia), secuestradores de ácidos biliares (colesevelam, Welcol), ácido bempedoico (Nexletol), icosapento etílico (Vascepa) o inhibidores de PCSK9 (evolocumab). Los pacientes que no pueden obtener el nivel requerido de LDL-C pueden requerir aféresis de LDL (equivalente a diálisis para la enfermedad renal).
Niños con hipercolesterolemia familiar heterocigótica (HeFH)
Al tratar a un niño con HF, los padres deben consultar con su médico. Cuando los niveles de LDL-C son superiores a 190 mg/dl, o superiores a 160 md/dl con al menos otros dos factores de riesgo presentes, se debe considerar el tratamiento. Según las pautas de la Asociación Nacional de Lípidos, se recomienda la derivación a un experto en lípidos, el control temprano de la dieta y la actividad física y la consideración de la medicación con estatinas. La FDA ha autorizado el uso de atorvastatina y rosuvastatina, dos de las estatinas más potentes, en niños, al igual que todas las estatinas menores.
El objetivo es una disminución del 50% en el LDL-C o LDL-C por debajo de 130 mg/dL. Las estatinas se pueden iniciar a los 8 o 10 años de edad; No ha habido informes de efectos secundarios de las estatinas en niños. Según los estudios, los niños que empiezan a tomar estatinas tienen una probabilidad estadísticamente significativamente menor de sufrir enfermedad de las arterias coronarias que sus padres afectados por HF. El propósito de comenzar a tomar estatinas en la infancia es disminuir la carga de exposición al C-LDL a lo largo de la vida.
Niños o adultos con hipercolesterolemia familiar homocigótica (HoFH)
Se sugiere tratamiento temprano y vigilancia con angiografía coronaria por TC y otras imágenes; Estos individuos frecuentemente requieren técnicas de tratamiento adicionales, ya que el tratamiento farmacéutico y las modificaciones en el estilo de vida pueden no ser suficientes. Las estatinas suelen recetarse tan pronto como se obtiene el diagnóstico (aunque pueden no ser efectivas como se explicó anteriormente). Lomitapida es ahora una terapia aprobada por la FDA para adultos con HoFH y debe investigarse en estos individuos, especialmente si los niveles de LDL-C son incontrolables con los medicamentos existentes.
- La FDA también aprobó un inhibidor de PCSK9 , evolocumab, para HoFH.
- Más recientemente, en 2021 , la FDA aprobó la inyección de Evkeeza (evinacumab-dgnb) como tratamiento complementario para pacientes de 12 años o más con HoFH.
- Las opciones adicionales incluyen aféresis de LDL o trasplante de hígado.
aféresis de LDL
La sangre se extrae de una vena a través de un catéter y se procesa para eliminar las partículas de LDL-C de forma similar a la diálisis renal . Los productos sanguíneos normales se devuelven utilizando un catéter diferente. Los niveles de LDL-C se reducirán aproximadamente un 50%, pero aumentarán entre los procedimientos de aféresis, por lo que se requieren semanalmente o cada dos semanas. La técnica es exitosa y bien tolerada, aunque requiere mucho tiempo y sólo se ofrece en 50 a 60 lugares en los Estados Unidos.
Trasplante de hígado
Los trasplantes de hígado son extremadamente raros y pueden serlo mucho más si se dispone de nuevos tratamientos. Debido a que el hígado del donante tiene receptores de LDL normales, el LDL-C pronto vuelve a la normalidad después del procedimiento, pero los riesgos de cualquier trasplante de órgano son altos, incluidas las complicaciones de una cirugía mayor y las consecuencias de la supresión del sistema inmunológico de por vida. Con frecuencia los órganos de donantes no están disponibles. Los pacientes con mutaciones patógenas familiares en los genes APOB o PCSK9 tienen receptores de LDL normales y no pueden someterse a un trasplante de hígado.
Las personas con HoFH pueden beneficiarse de modalidades de imágenes como ecocardiogramas, angiografías por tomografía computarizada y cateterismo cardíaco.
Conclusión
La HF es una enfermedad grave que se desarrolla en la primera infancia y tiene implicaciones a largo plazo. Aunque es bien reconocida la necesidad de un enfoque de detección temprana de esta enfermedad, no hay acuerdo sobre quién y cuándo realizar la prueba. El tratamiento hipolipemiante temprano y los cambios en el estilo de vida pueden mejorar los resultados clínicos. Si bien estos intentos de terapia han mejorado significativamente el pronóstico de la hipercolesterolemia familiar heterocigótica (HeFH), los resultados de la hipercolesterolemia familiar homocigótica (FH) siguen siendo insatisfactorios.
Aunque la mayoría de los casos pueden tratarse con una combinación de estatinas e inhibidores de la absorción de colesterol, algunos requerirán tratamientos más invasivos como la aféresis de LDL. En las últimas dos décadas, han surgido medicamentos innovadores para disminuir los niveles de colesterol LDL y posponer la aterosclerosis temprana, particularmente cuando se combinan con cambios en el estilo de vida. A pesar de estas victorias, la gran mayoría de los niños no alcanzan sus objetivos de lípidos debido a lagunas en el diagnóstico, el seguimiento y la terapia.