Craniopharyngioma
Overview
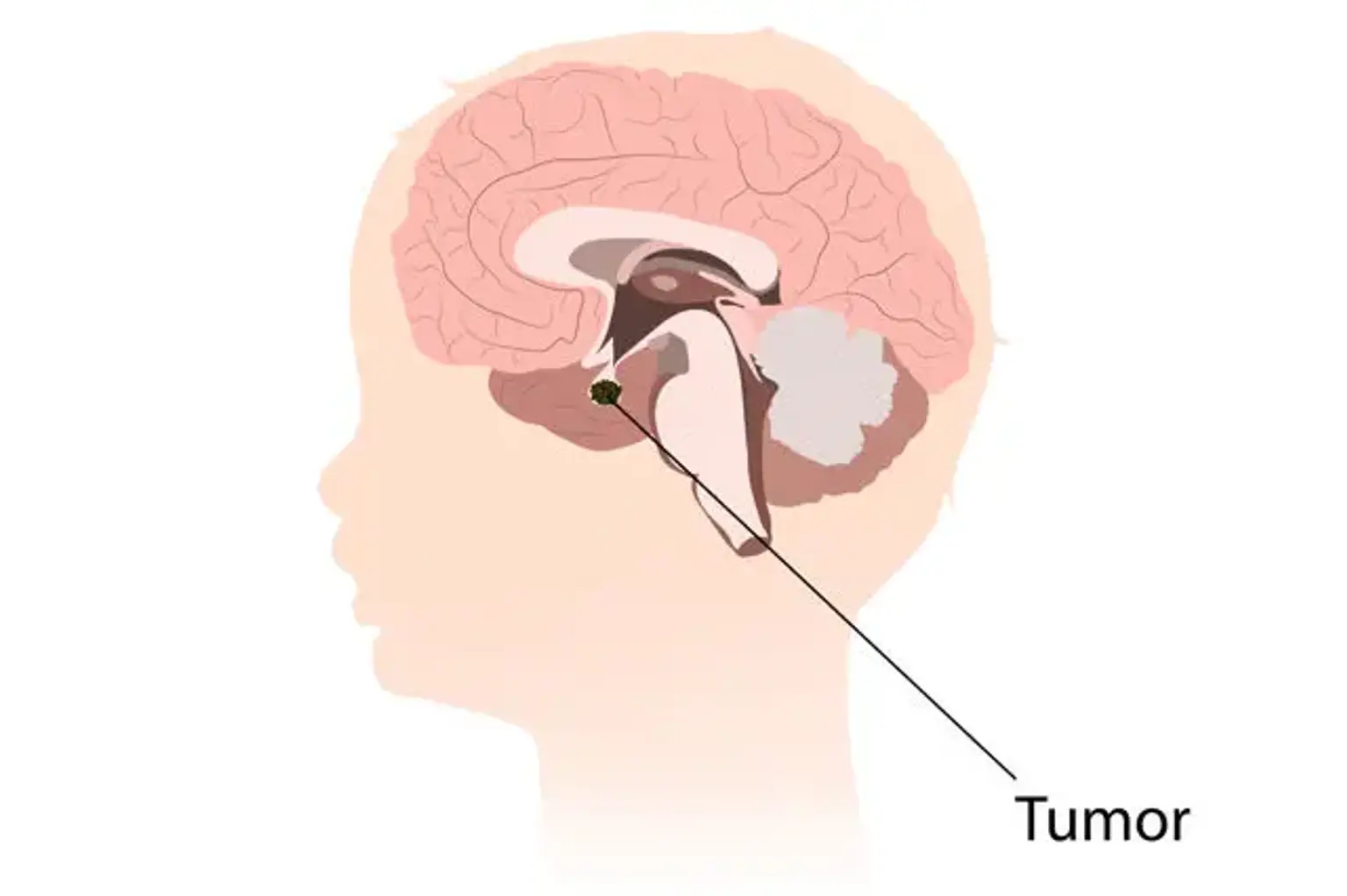
Craniopharyngioma is an uncommon, but harmless central nervous system tumor (CNS). It is a partially cystic embryonic abnormality that can arise in the sellar/parasellar area and cause a variety of symptoms including headaches, nausea and vomiting, visual problems, and endocrine disorders.
It poses a unique difficulty to the physicians who treat it, which often include neurosurgery, neuro-ophthalmology, neurology, endocrinology, and pediatrics. The difficulty stems from the tumor's capacity to attach to the surfaces around it. As a result, it is incredibly difficult to manage, and it is also infamous for its high recurrence rates.
Following histological evaluation of a surgical specimen, a definitive diagnosis is obtained. Endocrine disruptions are usually persistent and need cautious restoration. Overall, there is an 80% 5-year survival rate, albeit this is coupled with significant morbidity
Craniopharyngioma Definition
Craniopharyngiomas are slow-growing benign tumors that occur in the sellar and parasellar regions of the central nervous system. This tumor has a point prevalence of about 2/100,000. The development of symptoms is usually gradual, with most patients experiencing neurological (headaches, visual problems) and endocrine (growth retardation, delayed puberty) dysfunctions at the time of diagnosis.
Craniopharyngiomas are hypothesized to develop from epithelial remains of the craniopharyngeal duct or Rathke's pouch (adamantinomatous type) or from metaplasia of squamous epithelial cell rests that are vestiges of the stomadeum that contributed to the buccal mucosa (squamous epithelial cell rests) (squamous papillary type). The three components of the tumor (cystic, solid, and calcified) at the distinctive sellar/parasellar position are used to make the neuroradiological diagnosis.
Epidemiology
Craniopharyngioma occurs at a rate of 0.5 to 2 occurrences per million people each year. Craniopharyngiomas can develop at any age. Although it is typically thought to be a childhood condition, accounting for 1.2 to 4% of all brain tumors, roughly half of craniopharyngiomas are discovered in adults.
It exhibits a characteristic bimodal age distribution, with an enhanced incidence rate in children aged 5 to 14 years and in adults aged 50 to 74 years. There is no statistically significant variation in incidence depending on gender, race, or geographic area. Only two families have been known to have craniopharyngioma.
Craniopharyngioma has a very high recurrence rate of around 50%. It also has good survival rates (83 percent to 96 percent five-year survival and 65 percent to 100 percent 10-year survival), but it also has significant morbidity rates, with virtually all patients experiencing some sequelae.
Etiology
The embryonic hypothesis and the metaplastic theory are the two basic theories of craniopharyngioma formation. These two hypotheses correspond to the two histologic subgroups of craniopharyngiomas, namely adamantinomatous craniopharyngioma (ACP) and papillary craniopharyngioma (PCP)
The embryonic hypothesis is linked to the formation of ACP, a more prevalent subtype that can afflict people of all ages. The ectodermal lid of the stomodeum outpouches during development. This outpouching, known as Rathke's pouch, continues cranially towards the diencephalon's floor, eventually becoming the adenohypophysis, or anterior pituitary gland.
As it migrates cranially, its expansion generates the craniopharyngeal duct, which involutes later. Involution is not always complete, and remains of ectodermal cells might be found. These embryonic cells can multiply around the craniopharyngeal duct extension and develop into a craniopharyngioma.
Pathophysiology
The sellar/suprasellar area is the most prevalent site for craniopharyngioma, accounting for 95 percent of all craniopharyngiomas. Its pathophysiology is determined by its location. Craniopharyngiomas can compress normal pituitary tissue, leading to pituitary deficits, notably in anterior pituitary hormones.
It can also compress the optic chiasm and/or optic nerves, resulting in a variety of visual problems ranging from impaired vision to blindness. It can also cause hydrocephalus as a result of third ventricle compression. Non-specific intracranial hypertension symptoms such as headache, nausea, and vomiting can arise in situations of severe suprasellar extension. There have been reports of solitary oculomotor nerve and abducens nerve palsies.
The anterior circulation supplies blood to the majority of craniopharyngiomas. The main site of craniopharyngioma recurrence is the most prevalent, however metastatic foci may emerge as a result of seeding after surgery.
Craniopharyngioma Symptoms
Craniopharyngiomas are slow-growing tumors that are frequently detected late in the course of the patient's compressive symptoms. Symptoms often indicated the location of tumors and their closeness to nearby structures.
Headaches
Headaches are experienced by nearly half of all patients. Headaches may be caused by elevated intracranial pressure (ICP), which may be accompanied by nausea and vomiting, or by meningeal irritation caused by cystic fluid.
Visual Symptoms
Visual symptoms will be present in 62 to 84 percent of craniopharyngioma patients. Temporal hemianopsia caused by optic chiasm compression is the most frequent visual disturbance seen. The optic pathway is dysfunctional in 50-75 percent of individuals.
Hormonal Deficiency
- Growth hormone (GH) insufficiency, caused by the reduction in GH production - symptoms include:
- Stunted growth and delayed puberty in children
- General fatigue, loss of muscle mass and tone in adults
- Pituitary insufficiency
- Because craniopharyngiomas form in the region of the pituitary stalk, they can interfere with the function of the pituitary gland and many other hormones.
- Prolactin production reduction is extremely rare and only happens in acute pituitary insufficiency.
- Due to the "stalk effect," large pituitary tumors can paradoxically raise blood prolactin levels. This rise happens as a result of the pituitary stalk being compressed, which interferes with the brain's regulation of prolactin production.
- Elevated prolactin levels in premenopausal women might cause a decrease or lack of menstrual cycles and/or breast milk supply (galactorrhea).
- Prolactin levels are normally only modestly raised with stalk effect, as compared to prolactinomas, when prolactin levels are frequently quite high.
- Diabetes insipidus is caused by a lack of antidiuretic hormone, a posterior pituitary hormone. Among the symptoms are:
- Excessive thirst
- Excessive urination
- Adrenal insufficiency is caused by a decrease in ACTH production and a decrease in cortisol. It can be lethal in extreme circumstances. Among the symptoms are:
- Fatigue
- Low blood pressure
- Electrolyte abnormalities
Diagnosis
An endocrinologist, a neuro-opthalmologist, and a neurosurgeon must all be involved in the examination of a craniopharyngioma.
Imaging
During the assessment of visual problems, craniopharyngioma is identified using computed tomography (CT) and/or magnetic resonance imaging (MRI). MRI is the gold standard for diagnosing craniopharyngiomas and other pituitary tumors because it offers more information about the tumor, its location, and its relationship to the surrounding structures.
Both modalities show the cystic region, while CT imaging shows the most calcification. Craniopharyngioma has a diverse texture; the mix of solid, cystic, and calcification components on imaging modalities aids in diagnosis. ACP is typically huge irregular with 90% calcium and a cystic region, but PCP is mostly solid with just a few cysts and calcifications.
Visual Exam
In patients with visual abnormalities, a neuro-opthalmologist should do a comprehensive visual exam, including acuity and visual field.
Endocrine Evaluation
All patients should be tested for hormonal changes. Fasting morning cortisol, ACTH, TSH, free T4, follicle-stimulating hormone (FSH), estradiol (females), testosterone (males), GH, insulin-like growth factor-1, prolactin, serum sodium, and urine specific gravity and osmolality should all be tested. Dynamic testing, such as a cosyntropin stim test, can be used in individuals whose morning cortisol levels are ambiguous or uncertain.
Management
Because of their position, invasiveness, and closeness to surrounding neurovascular systems, craniopharyngiomas are difficult to treat. Surgery, radiation, and intracystic therapy are only a few of the treatment options for craniopharyngioma.
Each patient's therapy, operation type, and extent should be tailored to their specific needs. When deciding on a treatment plan, the patient's age, underlying medical co-morbidities, tumor location, kind, and invasiveness, and eventually neurosurgeon expertise should all be taken into account.
Residual tumor, verified by post-operative MRI, is typically treated with external beam radiation; however, stereotactic radiosurgery (gamma knife) has also been employed. The use of proton beam radiation for residual disease is being researched at the moment.
On rare occasions, a patient will appear with a completely cystic tumor. The stereotactic implantation of a catheter to allow repeated aspiration is also one of the treatment options for these tumors. In addition, intracystic radiation or chemotherapy (Bleomycin) has had some success.
There is no place for systemic chemotherapy, although immune treatment has lately been studied. When administered systemically, interferon alpha had little impact, although there has been considerable effectiveness when utilized intracystically.
Preoperative Management
Before surgery, hormone shortage, especially secondary adrenal insufficiency, and hypothyroidism should be addressed with glucocorticoid and thyroid hormone replacement. Hydrocortisone 20 to 30 mg in 2 to 3 separate doses or prednisone 5 to 10 mg in a single or divided dosage are used for glucocorticoid replacement. Levothyroxine is the most effective therapy to treat thyroid hormone deficiency.
To treat hydrocephalus, a temporary or permanent shunt may be required to drain cerebrospinal fluid.
Surgery
Surgical intervention is required to confirm the diagnosis, as well as to treat tumors that cause cognitive impairments, pituitary dysfunction, and hydrocephalus. Depending on the location of the craniopharyngioma, the most frequent surgical methods are endoscopic endonasal transsphenoidal (EET) or transcranial. It is debatable if resection should be extended. Gross complete resection has been linked to an increase in post-surgical impairments, but no change in recurrence rates.
Several grading systems have been created to assist in deciding on the optimal surgical modality depending on the location of the craniopharyngioma and its relationship to adjacent tissues. To plan the surgical approach, a categorization scheme based on the infundibulum is used.
Pre-infundibular tumors are sub-chiasmatic tumors that displac the optic chiasm superiorly and posteriorly. Trans-infundibular tumors of type II can extend into the third ventricle. Type III is retro-infundibular, and it can extend either superiorly or inferiorly into the pontine cisterns.
Type IV tumors are more commonly seen in the third ventricle. Except for type IV, ETT may be performed on all kinds. Craniotomy and transcallosal, transcortical, or trans-ventricular procedures are preferable for solitary third ventral tumors. ETT should be avoided in very big tumors that are mainly solid, have calcification, and are vascularly invasive.
Postsurgical Care
Patients should be closely monitored for cerebrospinal fluid (CSF) leakage following surgery.
Hormone deficit management is critical following surgery. Depending on the kind of operation and presurgical hormonal assessment, individuals in immediate postoperative care may require stress dosage steroids. The length of glucocorticoid medication can be determined by careful hormonal monitoring, such as measuring postoperative morning cortisol levels.
Monitoring sodium and urine osmolarity is critical for tracking the progression of diabetic insipidus. Thyroid hormone levels should be checked one to two weeks following surgery. At 3 months postoperatively, the sex hormone and GH levels can be measured. The amount of anterior pituitary hormone replacement varies from instance to case.
Radiotherapy
Radiation therapy is used to treat individuals who have residual illness or to avoid recurrences. External radiotherapy, proton beam treatment, stereotactic radiotherapy, radiosurgery, and brachytherapy are all types of radiation therapy. Radiotherapy's purpose is to reduce tumor burden while sparing critical neurological structures. For each radiation modality, a certain Gy dosage has been assigned.
Several studies have found that radiation treatment results in lower mortality and slightly lower morbidity. Regardless, radiation treatment has not been shown to lessen recurrence rates. As a result, it remains an adjuvant method to neurosurgical intervention.
It is commonly advised that radiation be administered following craniopharyngioma subtotal excision. This reduces the risk of the leftover tumor growing. However, there are long-term risks associated with radiation such as cataracts, aggravation of hypothalamic-pituitary dysfunction, cognitive dysfunction, radionecrosis.
In a recently published research, 20% of patients who had a sub-total resection remained stable without radiation during the follow-up period. At this time, it is unclear if all patients with residual tumors should get immediate post-operative radiation.
There is evidence that surgical expertise influences the clinical outcome of craniopharyngioma patients. The sub-total excision of a tumor infiltrating the hypothalamus is difficult, however it may be hypothesized that the smaller the remaining component, the better the efficacy of post-operative radiation.
Despite the use of radiation for residual illness, roughly 20% of patients will have the disease relapse. In many cases, aggressive tumor removal is the only choice. The long-term clinical prognosis of these individuals is unknown at this time.
Intracystic Therapy
Intracystic treatment is most commonly utilized to treat solely cystic craniopharyngioma. In intracystic treatment, toxic chemicals such as radioactive isotopes, bleomycin, and interferon-alpha are utilized to induce tumor fibrosis and sclerosis. Although this approach has been shown to result in considerable cyst shrinking, there is insufficient evidence on its application and support. One downside of this method is that severe neurotoxicity might occur in some patients because to sclerosing material cystic leaking.
Historical treatment of patients with Craniopharyngiomas
The therapy of individuals with craniopharyngiomas in the past has been described as a pendulum. The gross complete excision of a tumor infiltrating the hypothalamus is technically difficult yet doable. There is, however, a clear mortality rate (up to 10%) with this treatment, as well as a recurrence rate despite a gross complete resection (up to 15 percent(
The clinical outcome, however, can be less than ideal, with hypothalamic dysfunction (hyperphagia, obesity, behavioral disorders, memory problems, loss of neurovegetative homeostasis) and an altered neuropsychological profile (marked distractibility, difficulties in perceptual organization, poor verbal memory) having a significant impact on daily activities in both adult and paediatric patients, even with hormonal replacement therapy.
As a result, the pendulum has swung away from trying massive complete resection in patients with hypothalamic invasion and toward a less aggressive strategy. The hypothalamus component of the tumor is left in these individuals and treated with post-operative radiation.
There is some recent evidence in a paediatric population that the 5-year survival rate in patients with this dichotomized regime is 80%, which is consistent with previously published series in which all patients underwent a gross total resection. While there is no change in mortality with this treatment procedure, there is some indication that this cohort has less hypothalamic dysfunction.
Differential Diagnosis
The differential diagnosis may be considered under four main headings :
- Congenital anomalies
Arachnoid cyst and Rathke's cleft cyst.
- Other Tumours
Pituitary tumour, metastasis, meningioma, epidermoid and dermoid tumour, hypothalamic-optic pathway glioma, hypothalamic hamartoma, teratoma.
- Infectious/Inflammatory processes
Eosinophilic granuloma, lymphocytic hypophysitis, sarcoidosis, syphilis and tuberculosis.
- Vascular malformations
Aneurysm of the internal carotid or anterior communicating artery, arterio-venous malformation.
Prognosis
The prognosis of craniopharyngiomas is determined by the tumor's size, histologic type, surgical technique, and the degree of hypothalamic and endocrine deficiency. At 5 years, the overall survival rate of craniopharyngiomas is 80 to 95 percent. Young people have a fair prognosis, whereas those over the age of 65 have a bad prognosis. Females are more likely to have increased morbidity, such as stroke, unfavorable cardiac events, and psychological problems.
Malignant Craniopharyngioma
Craniopharyngiomas are often treated satisfactorily with adjuvant chemotherapy and neurosurgery. According to recent study, malignant changes of these ordinarily benign tumors are quite rare. Malignant craniopharyngiomas can arise at any age, are somewhat more prevalent in females, and are often adamantinomatous in nature.
Malignant transformations can take years to occur necessitating a longer follow-up period in patients diagnosed with the more prevalent benign variants. There was no relationship established between malignancy and first chemoradiotherapy treatment, and overall survival was quite low, with the median survival being 6 months following malignancy diagnosis.
Prevention
Although the origins of craniopharyngioma are unknown, it can occur in both children and adults, with a peak incidence occurring between the ages of 9 and 14 years. In the United States, around 120 cases are identified each year in individuals under the age of 19.
More than half of all craniopharyngioma patients are under the age of 18. There is no obvious link between the tumor and a certain gender or race. Craniopharyngiomas do not appear to "run in families" or to be passed down from parents.
Conclusion
Craniopharyngiomas are uncommon benign central nervous system cancers (CNS). These individuals typically come to their primary care practitioner with a variety of symptoms, including headaches, nausea and vomiting, visual problems, and endocrine changes. It poses a unique set of challenges for the clinicians who treat it, who often include neurosurgeons, neuro-ophthalmologists, neurologists, endocrinologists, and pediatricians.
Specialty trained nurses in anesthesia, operating rooms, and critical care are required to provide care. For the best results, an interprofessional team is required. The difficulty stems from the tumor's capacity to attach to the surfaces around it. As a result, it is incredibly difficult to manage, and it is also infamous for its high recurrence rates. However, the majority of people's results are positive.