Familial hypercholesterolemia
Overview
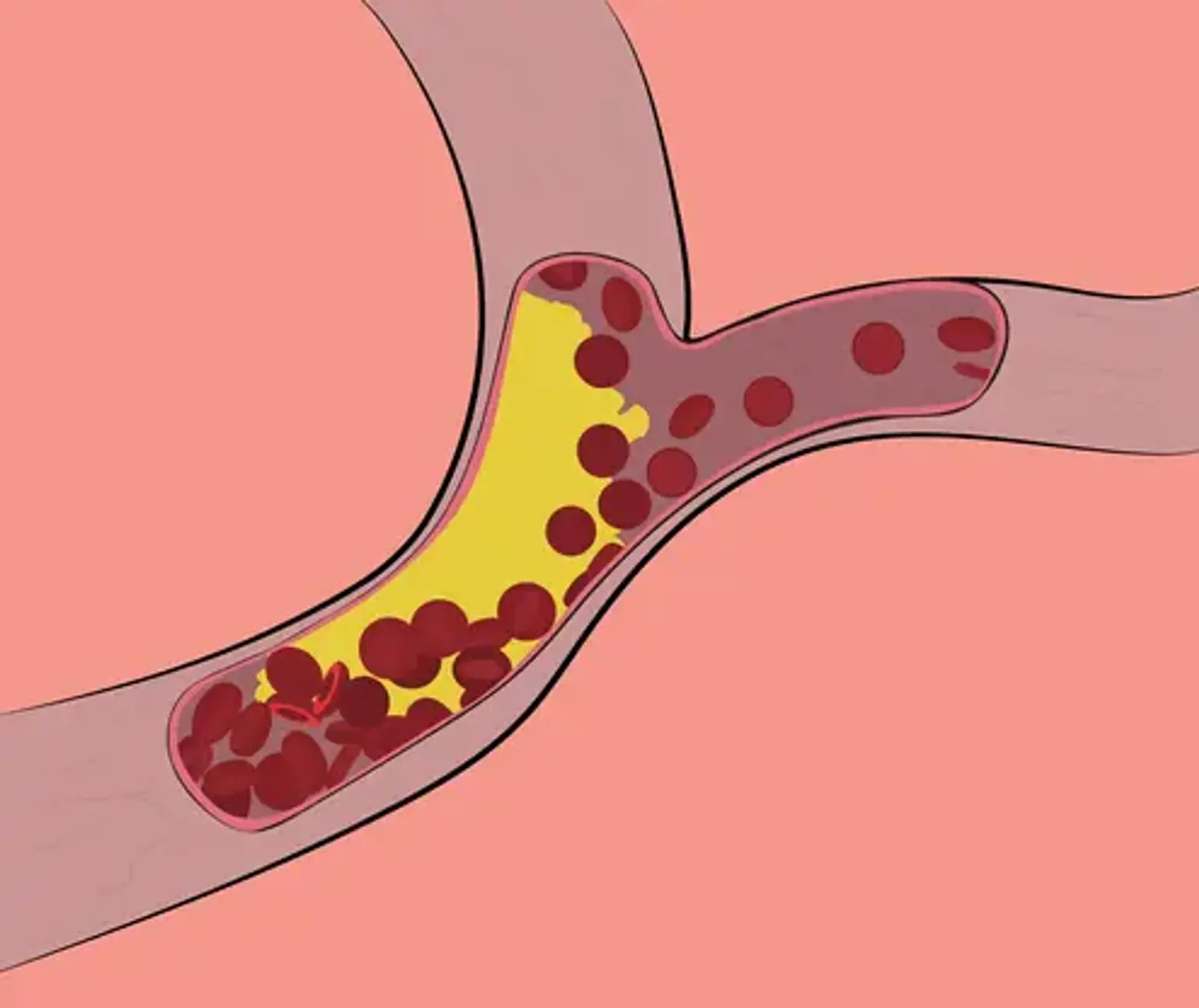
Familial hypercholesterolemia is a collection of inherited genetic abnormalities that cause a significant increase in blood cholesterol level. Although FH atherosclerosis develops largely in adulthood, it can begin as early as the first decade of life. The fact that early treatment of risk factors can reverse atherosclerotic alterations in the vascular system emphasizes the need of detecting and treating children with this disease as soon as possible.
What is Familial hypercholesterolemia?
Familial hypercholesterolemia (FH) is a genetic disease that affects how the body recycles LDL (bad) cholesterol. As a result, LDL levels in the blood remain extremely high; in severe instances, levels can exceed 190 milligrams per deciliter (mg/dL)
People who have FH are born with elevated LDL cholesterol. Everyone's cholesterol levels tend to grow as they become older. However, patients with FH have LDL levels that begin high and rise with time.
The last decade has seen a surge in study into this disease's genetic origin and treatment. However, because of a lack of understanding among pediatricians and the general public, FH is frequently misdiagnosed until the irreversible effects of atherosclerosis have been established.
This, like non-inherited cholesterol abnormalities, adds to atherosclerotic plaques, putting you at a significantly increased risk of coronary heart disease. People with FH are 20 times more likely to develop heart disease if they are not treated.
Men with FH develop coronary heart disease 10 to 20 years sooner. Before the age of 50, half of men with untreated FH will have a heart attack or angina. Some will be in their early twenties. Coronary heart disease can occur up to 20 to 30 years sooner in women. Approximately 30% of untreated women will have a heart attack before the age of 60.
These higher risks are unrelated to other risk factors, which might exacerbate the situation. The good news is that FH can be controlled with a mix of lifestyle modifications and medicines.
How frequent is Familial Hypercholesterolemia (FH)?
According to recent research, FH is as frequent as 1 in 250 people, making it one of the most common hereditary illnesses. However, given their significant risk, the majority of people go misdiagnosed and undertreated.
It is believed that 1 in 1/160,000 to 1 250,000 people have familial hypercholesteremia (FH). FH is more likely to develop in nations with a high frequency of FH, particularly those with a high rate of consanguinity (marriage between cousins).
Related Disorders
FH can be diagnosed using any of the established standardized diagnostic criteria listed in the "Diagnosis" section. When a diagnosis of FH is suspected but cannot be completely confirmed based on the available evidence, genetic testing can frequently help. In 2019, an international expert panel issued a consensus document in favor of the value of FH genetic testing, as well as a declaration stating that genetic testing should be administered as standard of care.
When genetic testing is negative, the technological limits and sensitivity of FH genetic testing cannot definitively rule out FH but may promote the examination of alternate reasons for the presenting symptoms. There are other illnesses that have comparable laboratory results or family history aspects to FH. These are some examples:
- Obesity, diabetes mellitus, hypothyroidism, medicines such as steroids, or renal disease can all cause hypercholesterolemia. The pattern of inheritance is non-Mendelian.
- Polygenic hypercholesterolemia is caused by a slew of minor impact genetic variations in a variety of cholesterol-metabolizing genes. Risk factors such as diabetes and obesity can aggravate polygenic hypercholesterolemia. An individual may have a familial history of hyperlipidemia and coronary artery disease, even if the onset of CAD occurs later in life. Inheritance follows a non-Mendelian pattern, and family members frequently appear with varying expression of LDL-C levels or cardiovascular risks.
- Lipoprotein a (Lp) is a cholesterol-like particle that, when increased (>30 mg/dL), increases the risk of early coronary artery disease (CAD). Lipoprotein levels are mostly influenced by genetics and are inherited in an autosomal codominant manner. Individuals with highly raised Lipoprotein levels may have a family history that resembles that of someone with FH, even if their LDL-Cholesterol levels are normal. Individuals who have both FH and excessive Lipoprotein are at a higher risk of developing early coronary artery disease.
Pathophysiology of FH
The phenotypic expression of faulty lipoprotein metabolism induced by a number of genetic disorders is known as familial hypercholesterolemia. Following Brown and Goldstein's important finding that mutations in the low-density lipoprotein receptor (LDLR) were the cause of monogenetic FH, approximately 1,500 mutations in this gene have been identified, accounting for more than 80% of instances of monogenetic FH.
Low-density lipoprotein (LDL) carries the bulk of plasma cholesterol through binding to the LDLR cell membrane via two ligands on LDL, apolipoprotein B-100 (apoB-100) and apoE. The LDL/LDLR combination penetrates cells, then LDL is released and LDLR recycles to the cell membrane.
The liver secretes the proprotein PCSK9, which binds to LDLR extracellularly and prevents its recycling to the cell membrane. As a result, PCSK9 decreases LDLR while increasing serum LDL. The final effect of these three major gene mutations is a binding malfunction of LDL receptors to LDL cholesterol, resulting in a decrease in LDL cholesterol absorption and destruction in the liver and an increase in blood LDL levels. Clinically, it is impossible to identify the underlying gene mutation in familial hypercholesterolemia.
Familial Hypercholesterolemia Symptoms
A thorough family history is required for the evaluation and diagnosis of familial hypercholesterolemia. First-degree relatives with a family history of early coronary artery disease or high cholesterol (younger than 60 years in women and 55 years in males) are warning flags. It should also raise the possibility of familial hypercholesterolemia in young individuals who have second-degree relatives who have had early coronary episodes.
Familial Hypercholesterolemia types
- Homozygous Familial Hypercholesterolemia
These individuals have ischemic heart disease, peripheral vascular disease, cerebrovascular illness, or aortic stenosis symptoms. Tendonitis or arthralgia may be present, as well as a history of odd skin lesions. Survival after the age of 30 is difficult unless unconventional procedures are used.
- Heterozygous Familial Hypercholesterolemia
Since childhood, these individuals have had significant hypercholesterolemia. Ischemic heart disease symptoms are widespread, especially when additional cardiovascular risk factors are present. Recurrent Achilles tendonitis or arthritic symptoms may be present.
Physical Examination
Typically, abnormal physical tests are associated with cholesterol depositions in the eye or skin. With homozygous familial hypercholesterolemia, it can occur at a young age. Tendon xanthomas, which appear as thickening of the tendons due to cholesterol accumulated among macrophages in connective tissue, are pathognomonic for familial hypercholesterolemia. The Achilles tendon and finger extensor tendons are the most commonly affected, but patellar and triceps tendons are also prevalent.
Tendon xanthomas affect fewer than half of FH patients. Corneal arcus senilis are cholesterol depositions surrounding the corneal rim. It is more specific in patients under the age of 45. Cholesterol deposition can also take the form of yellow-orange tuberous xanthomas (on the palms, elbows, or knees) or xanthelasma (in the eyelids), which are more specific for FH in individuals aged 20 to 25 years.
Diagnosis of Familial Hypercholesterolemia
Untreated children with LDL-C levels above 160 mg/dL, or with LDL-C levels above 130 mg/dL with a positive family history of FH or premature cardiac disease, should be evaluated for FH. An LDL-C exceeding 190 mg/dL, a personal and/or family history of early CAD, physical indications such as those indicated under "Symptoms," or a relative known to have FH should raise the suspicion of FH in untreated adults.
An FH variation is discovered roughly 60-80 % of the time in persons suspected of having FH based on clinical criteria. Although DNA testing is considered the "gold standard," it is not always required or possible. When it is unclear whether an individual is impacted or not, DNA testing should be explored, and it is especially useful for testing family members. Recent research also suggests that individual risks for CAD differ depending on the afflicted gene and kind of DNA mutation.
Individuals with an LDL-C >190 mg/dL and an FH pathogenic variation have a tenfold greater relative risk of CAD (compared to the general population), whereas those with an LDL-C >190 mg/dL but no FH pathogenic mutation have a threefold increased relative risk of CAD (compared to the general population). Individuals who test positive on genetic testing may have a higher risk of CAD than those who test negative on genetic testing at any given LDL-C level.
Once an individual has been diagnosed with FH, a process known as "cascade screening," "cascade testing," or "family screening" (testing of close relatives) is recommended to identify those with FH before symptoms appear, allowing for early and intensive treatment to begin and disease and death to be avoided. If a pathogenic variation is found, risk in the patient's first degree relatives (parent, sibling, child) and, if necessary, more distant relatives can be determined by DNA testing by tracking the changed gene through the family.
If DNA testing is not conducted, a cholesterol-based variation of cascade screening can be used. Cascade screening, by any method, has been demonstrated to be useful in identifying individuals with FH who were not being treated adequately. A genetic counselor can assist a family with this procedure.
Clinical Testing and Workup
Evaluations following initial diagnosis
The following assessments are indicated in adults and children to determine the amount of illness and requirements of an individual diagnosed with FH:
- Lipid levels before therapy
- Lipoprotein(a) levels should be measured whenever feasible since lipoprotein(a) is an additional risk factor for CAD.
- Exclusion of concomitant conditions that might impact lipid readings (kidney disease, untreated hypothyroidism, acute myocardial infarction, infection).
- Total cholesterol (TC), low density lipoprotein cholesterol (LDL-C), high density lipoprotein cholesterol (HDL-C), and triglycerides are all part of the lipid panel.
- Consultation with a lipid specialist or physician with knowledge in FH Medical genetics
Some guidelines propose noninvasive imaging techniques (e.g., assessment of carotid intima-media thickness) in children to assist inform treatment options.
Management
FH treatment focuses on lowering LDL-C levels in order to reduce the risk of atherosclerotic heart disease.
Adults with FH
- Lifestyle intervention
Dietary adjustments such as limiting saturated fat and removing trans fats have a major influence on cholesterol levels. Reduced dietary cholesterol and increased soluble fiber are also beneficial. Vegetables, whole fruit and grains, nuts and legumes should be the mainstays of the diet. The best sources of animal protein include seafood, lean chicken, and low-fat dairy products. Weight reduction and aerobic exercise have very little impacts on cholesterol levels, but they can help decrease blood pressure and blood sugar levels, and hence the risk of cardiovascular disease.
- Cholesterol lowering medication
Adults should begin therapy as soon as possible after being diagnosed. Almost all will require cholesterol-lowering medication. A positive FH diagnosis should prompt more intensive therapy than would ordinarily be given to a patient with "garden type" high cholesterol. Some guidelines recommend that untreated cholesterol levels be decreased by at least 50%; others recommend that those without a past CVD incident aim for fewer than 100 mg/dL. When other risk factors such as diabetes or atherosclerosis are present, LDL-C objectives are more severe (usually 70 mg/dl). If these goals cannot be met in the primary care context, patients with FH should be referred to a lipidologist.
Pharmacotherapy should begin with statins, followed by the inclusion of additional medicines if the desired LDL-C level is not obtained with statins and lifestyle adjustments. One of the higher potency statins (atorvastatin or rosuvastatin) administered at the maximum dose should be preferred.
Muscle damage (rhabdomyolysis) is the most common side effect of statins, yet it occurs in only around 1 in 10,000 people who take them. Damage to the liver does not occur at a greater incidence in statin users than in non-statin users. Myalgia (muscle discomfort) is a moderately frequent side effect that affects 10-15% of people. Minor myalgia, with or without mild creatine kinase elevations (less than 5 times the upper limit of normal), is not always cause to stop using statins or other cholesterol-lowering drugs.
Other medications may be required, such as ezetimibe (Zetia), bile acid sequestrants (colesevelam, Welcol), bempedoic acid (Nexletol), icosapent ethyl (Vascepa), or PCSK9 inhibitors (evolocumab). Patients who are unable to obtain the required LDL-C level may require LDL apheresis (equivalent to dialysis for renal disease).
Children with Heterozygous Familial Hypercholesterolemia (HeFH)
When treating a kid with FH, parents should consult with their doctor. When LDL-C levels are more than 190 mg/dl, or greater than 160 md/dl with at least two other risk factors present, treatment should be considered. According to the National Lipid Association guidelines, referral to a lipid expert, early management of diet and physical activity, and consideration of statin medication are all recommended. The FDA has authorized the use of atorvastatin and rosuvastatin, two of the stronger statins, in children, as have all of the lesser statins.
The target is a 50% decrease in LDL-C or LDL-C below 130 mg/dL. Statins can be started as early as 8 to 10 years old; there have been no reports of statin side effects in children. According to studies, children who start taking statins have a statistically significant lower chance of getting coronary artery disease than their FH-affected parents. The purpose of starting statins in childhood is to lessen the lifelong burden of LDL-C exposure.
Children or Adults with Homozygous familial hypercholesterolemia (HoFH)
Early treatment and surveillance with CT coronary angiography and other imaging is suggested; these individuals frequently require additional treatment techniques, since pharmaceutical treatment and lifestyle modifications may not be enough. Statins are often prescribed as soon as a diagnosis is obtained (though may not be effective as explained above). Lomitapide is now an FDA-approved therapy for adults with HoFH and should be investigated for these individuals, especially if LDL-C levels are uncontrollable with existing medications.
- A PCSK9 inhibitor, evolocumab, was also approved by the FDA for HoFH.
- Most recently, in 2021, the FDA approved Evkeeza (evinacumab-dgnb) injection as an add-on treatment for patients aged 12 and older with HoFH.
- Additional options include LDL apheresis or liver transplantation.
LDL apheresis
Blood is extracted from a vein through a catheter and processed to eliminate LDL-C particles in a manner similar to renal dialysis. Normal blood products are returned using a different catheter. LDL-C levels will reduce by around 50% but will increase between apheresis procedures, thus they are required weekly or every other week. The technique is successful and well tolerated, although it is time-consuming and only offered in 50-60 locations in the United States.
Liver transplantation
Liver transplants are extremely rare and may become much more so if new treatments become available. Because the donor liver has normal LDL receptors, the LDL-C soon returns to normal following the procedure, but the hazards of any organ transplant are high, including complications from major surgery and the consequences of lifetime immune system suppression. Donor organs are frequently unavailable. Patients with family pathogenic APOB or PCSK9 gene mutations have normal LDL receptors and cannot undergo liver transplantation.
Individuals with HoFH may benefit from imaging modalities such as echocardiograms, CT angiograms, and cardiac catheterization.
Conclusion
FH is a serious disease that develops in early childhood and has long-term implications. Although the necessity for an early detection approach for this disease is well acknowledged, there is no agreement on who and when to test. Early lipid-lowering treatment and lifestyle changes may enhance clinical outcomes. While such therapy attempts have significantly improved the prognosis of Heterozygous Familial Hypercholesterolemia (HeFH), the results of familial homozygous hypercholesterolemia (FH) remain unsatisfactory.
Although the majority of cases may be treated with a combination of statins and cholesterol absorption inhibitors, some will require more invasive treatments such as LDL apheresis. Over the last two decades, there has been an emergence of innovative medicines to decrease LDL cholesterol levels and postpone early atherosclerosis, particularly when combined with lifestyle changes. Despite these victories, the vast majority of children do not meet their lipid targets due to gaps in diagnosis, monitoring, and therapy.