Neuromuscular Diseases
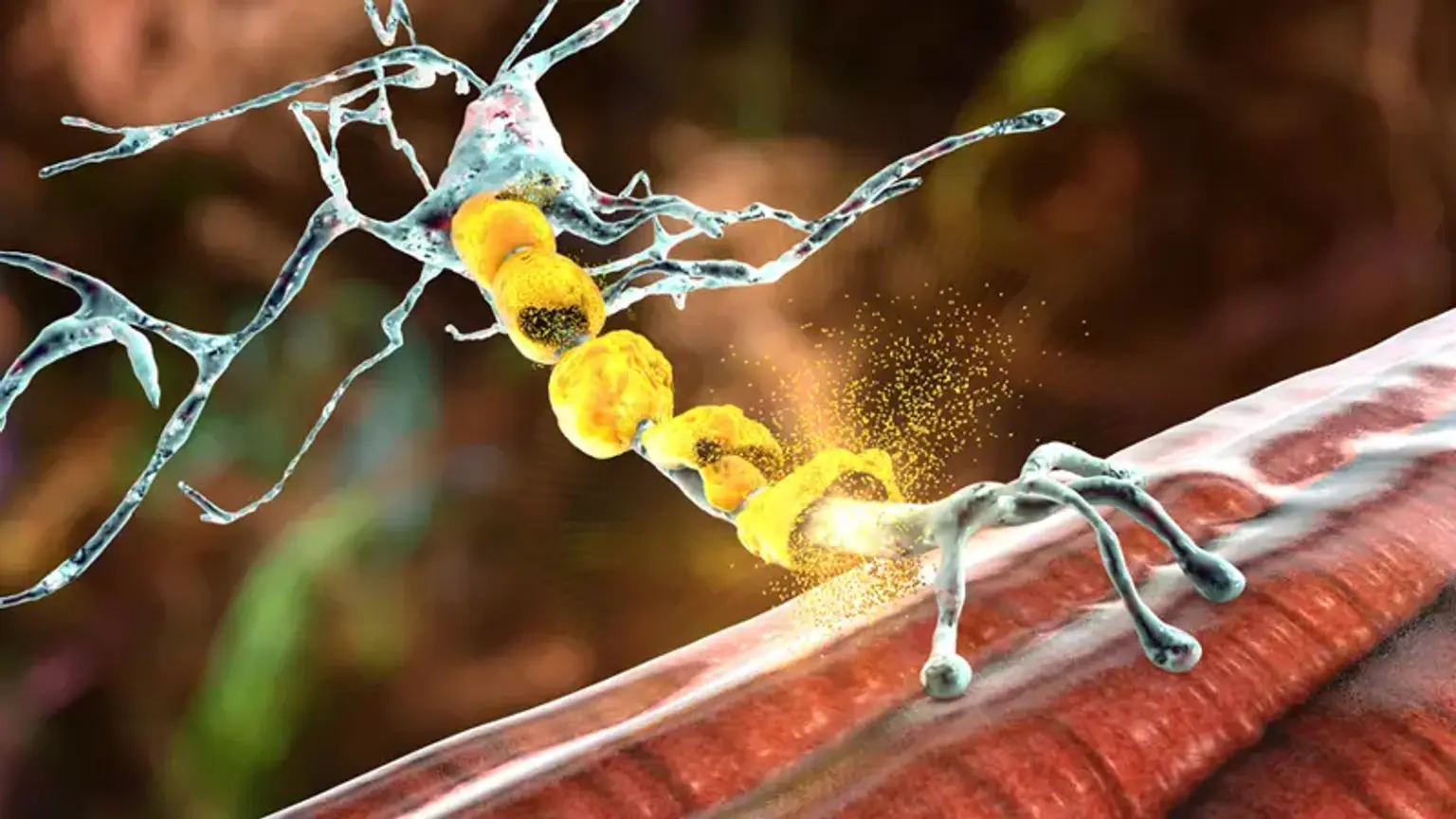
Overview
The neuromuscular system is the combination of the neurological system and muscles that work together to allow movement. Through specialized nerves, the brain directs the motions of skeletal (voluntary) muscles. The neuromuscular system is the combination of the neurological system and muscles that work together to allow movement.
A message is transmitted to certain neurons (nerve cells) called higher motor neurons when you desire to move a section of your body. Upper motor neurons have lengthy axons that go into and through the brain, as well as into the spinal cord, where they link with lower motor neurons. Lower motor neurons in the spinal cord deliver their axons directly to the muscle they regulate via nerves in the arms and legs.
There are several illnesses classed as neuromuscular disorders. Neuromuscular diseases induce muscle weakness in the body as a result of a breakdown in communication between the neurological system and the muscles it controls.
Neuromuscular Disorders Definition
Neuromuscular disorders are a group of illnesses that affect the peripheral nervous system, which includes all of the motor and sensory nerves that connect the brain and spinal cord to the rest of the body. The most common symptom of these illnesses is progressive muscular weakening. The injured location might be anywhere.
- The cell bodies (i.e., Amyotrophic lateral sclerosis [ALS] or sensory ganglionopathies),
- Axons (i.e., axonal peripheral neuropathies or brachial plexopathies),
- Schwann cells (i.e., chronic inflammatory demyelinating polyradiculoneuropathy),
- Neuromuscular junction (i.e., myasthenia gravis or Lambert-Eaton myasthenic syndrome),
- Muscle (i.e., inflammatory myopathy or muscular dystrophy), or any combination of these sites.
Some neuromuscular illnesses, such as ALS, are also associated with central nervous system disease, although the majority are limited to the peripheral nervous system.
Neuromuscular disorders Types
Types of neuromuscular disorders include:
- Amyotrophic lateral sclerosis (ALS)
- Charcot-Marie-Tooth disease
- Multiple sclerosis
- Muscular dystrophy
- Myasthenia gravis
- Myopathy
- Myositis, including polymyositis and dermatomyositis
- Peripheral neuropathy
- Spinal muscular atrophy
Myasthenia gravis (MG)
Myasthenia gravis (MG) is the most frequent condition affecting the skeletal muscles' neuromuscular junction (NMJ). The characteristic symptom is a fluctuating weakness that worsens in the afternoon. It often affects the muscles of the eyes, throat, and extremities. Muscle weakness results from the decreased passage of electrical impulses across the neuromuscular junction caused by the development of autoantibodies against certain postsynaptic membrane proteins.
Myasthenia gravis is associated with a large range of problems. These include myasthenic crises, an acute respiratory paralysis that need urgent care, as well as long-term medication-related side effects such as opportunistic infections and lymphoproliferative cancers.
Myasthenia gravis Causes
Myasthenia gravis, like other autoimmune illnesses, affects those who are genetically predisposed. Infections, vaccines, operations, and medicines are examples of precipitating causes.
Myasthenia Gravis Classification
MG can be categorized into several subgroups based on the clinical characteristics and antibodies involved. Each group reacts differently to therapy and so has prognostic value:
- Early-onset MG: Age at onset less than 50 years with thymic hyperplasia
- Late-onset MG: Age at onset greater than 50 years with thymic atrophy
- Thymoma-associated MG
- Ocular MG: Symptoms only from periocular muscles
- MG with no detectable AChR and MuSK antibodies
Myasthenia gravis symptoms
The distinguishing clinical feature of MG is the fluctuating muscle weakness that varies in severity, worsens with physical activity, and improves with rest. It can be precipitated by a wide variety of factors like
- Infections
- Surgery
- Immunization
- Heat
- Emotional stress
- Pregnancy
- Drugs (commonly aminoglycosides, fluoroquinolones, beta-blockers, neuromuscular blocking agents), and
- Worsening of chronic medical illnesses.
Patients should be asked about the timing of their symptoms, when time of day they normally occur, and how they improve with rest throughout the history taking process. Inquire about minor symptoms such as coughing after swallowing, taking longer to complete eating, hoarseness of voice, easy fatiguability in ascending upstairs, and sluggish and frequent errors in writing or typing; these symptoms are particularly noticeable towards the end of the day or work shift.
The most common symptoms include the following:
- Extraocular Muscle Weakness: On the initial presentation, around 85 percent of patients will have this. Diplopia, ptosis, or both are common patient complaints. Within two years, 50 percent of patients would develop widespread MG encompassing the bulbar, axial, and limb muscles.
- Bulbar Muscle Weakness: This is the first symptom in 15% of patients and produces symptoms such as difficulty chewing or frequent choking, dysphagia, hoarseness, and dysarthria. Facial muscle involvement results in an expressionless face, whereas neck muscle involvement results in dropped-head syndrome.
- Limb Weakness: The proximal muscles are generally more afflicted than the distal muscles, with the upper limbs being more impacted than the lower limbs.
- Myasthenic crisis: It is a medical emergency caused by the involvement of the intercostal muscles and diaphragm.
Because MG exclusively affects nicotinic cholinergic receptors, there are no autonomic symptoms such as palpitations, bowel or bladder difficulties.
Because of the variable illness pattern, a physical examination may demonstrate normal muscular strength. Repeated or prolonged muscular contractions might reveal weakness in such instances. Rest or application of ice (ice-pack test) to the affected muscle group results in improvement. Normal pupils, deep tendon reflexes, and sensory exams
Myasthenia gravis Diagnosis
The majority of MG diagnoses are clinical. Typically, laboratory tests and techniques assist the doctor in validating clinical findings.
- Serologic Tests:
The anti-AChR Ab test is very specific and confirms the diagnosis in individuals with traditional clinical symptoms. It affects four-fifths of people with generalized MG but only half of those with pure ocular MG.
- Electrophysiologic Tests:
These are important in patients who test negative for antibodies. The repeated nerve stimulation (RNS) test and single-fiber electromyography are two commonly used tests for MG. Both tests examine the NMJ for conduction delays. Before doing these tests, routine nerve conduction investigations are routinely conducted to evaluate the function of the nerves and muscles.
- Edrophonium (Tensilon) Test:
Edrophonium is an acetylcholinesterase inhibitor with a short half-life that enhances ACh availability in the NMJ. This is especially important for ocular MG, where electrophysiologic testing is not possible. It is given intravenously, and the patient's symptoms of ptosis or diplopia are monitored for improvement. It has a sensitivity range of 71% to 95% for MG diagnosis.
- Ice-pack Test:
When edrophonium testing is not possible, an ice-pack test can be used. This test necessitates the application of an ice pack to the eye for 2-5 minutes. Then, any improvements in ptosis are evaluated. This test cannot be used to assess extraocular muscles.
- Imaging:
Patients with MG should have a chest computed tomography (CT) or magnetic resonance imaging (MRI) to rule out thymoma. In cases of pure ocular MG, an MRI of the orbits and brain is required to rule out any localized mass lesions.
Myasthenia gravis Treatment
Cholinesterase enzyme inhibitors and immunosuppressive medicines are the mainstays of MG therapy. Plasmapheresis or intravenous immunoglobulins can be utilized to treat symptoms that are resistant to initial treatment methods or those that require quick remission of symptoms .
Management strategies in MG are based on the following four principles:
- Symptomatic Treatment:
Acetylcholinesterase inhibitors raise ACh levels at the NMJ by inhibiting enzymatic breakdown. Because of its extended duration of action, pyridostigmine bromide is favored over neostigmine. Ambenonium chloride can be utilized in those who have a bromide intolerance that causes gastrointestinal symptoms. Patients with MuSK MG have a poor response to these medications and may require greater doses.
- Immunosuppressive Treatment:
These are prescribed for people who continue to experience symptoms despite receiving pyridostigmine medication. The first-line immunosuppressive medications used in the therapy of MG include glucocorticoids (prednisone, prednisolone, and methylprednisolone) and azathioprine. Cyclosporine, methotrexate, mycophenolate, cyclophosphamide, and tacrolimus are examples of second-line agents.
When a patient is resistant to therapy, has a contraindication to treatment, or is intolerable to the use of first-line medications, these are utilized. Various monoclonal antibodies, including rituximab and eculizumab, have recently been used to treat drug-resistant MG, although clinical trial evidence on their effectiveness is still lacking.
- Intravenous immunoglobulins (IVIG) / Plasmapheresis:
This is used to stabilize a patient before an operation during the perioperative phase. It is also the therapy of choice for myasthenic crises due to its early start of action and is utilized in circumstances when immunosuppressive medications have failed.
Charcot-Marie-Tooth disease
Inherited peripheral neuropathies are a set of illnesses that include hereditary motor and sensory neuropathies (HMSN), hereditary motor neuropathies (HMN), and hereditary motor neuropathies (HMN) (HMN). HMSN, the most prevalent entity, is also known as Charcot-Marie-Tooth illness (CMT).
Normal peripheral nerve organization and function are dependent on the intimate anatomical and physiological connection between Schwann cells and axons. Schwann cells' survival, proliferation, and differentiation are all determined by their axons. These cells, in turn, play a vital role in ion channel regulation as well as axon maintenance, survival, and regeneration. Primary demyelination and axonopathy are caused by mutations in genes that affect myelin assembly and axonal transport.
Charcot-Marie-Tooth disease symptoms
Distal symmetrical weakness, wasting, hypo/areflexia, and skeletal abnormalities are clinical symptoms shared by all CMTs. These are more noticeable in the lower limbs than in the upper limbs. Symptoms commonly mentioned include difficulties walking or running quickly, stumbling, falling, and ankle twisting or spraining. Motor milestones may be delayed in patients.Patients develop foot drop and a high-stepping stride as their weakness worsens.
Hand weakness presents as difficulties buttoning, zipping, and writing. Upper limb onset and proximal muscle weakness are also possible, although rare. Sensory sensations and indications are less noticeable. Around 20% to 30% of patients report pain, which is usually musculoskeletal and very rarely neuropathic. As a result, paresthesia and positive sensory sensations are uncommon.
In individuals with long-standing illness, examination reveals pes cavus, hammertoes, and clawed hands due to intrinsic muscular weakening. Leg and distal thigh waste may appear to be an inverted champagne bottle. Spinal abnormalities (scoliosis) are also possible. Proprioceptive loss and bone abnormalities such as pes cavus and hammertoes induce abnormal gait and instability.
The onset age spans from infancy to the elderly, with the majority occurring within the first two decades of life. Because the beginning is gradual and the development is generally modest, determining the precise age of onset can be challenging.
The severity of the handicap varies from patient to patient, ranging from mild to severe. Patients differ in their age of start, motor impairments, and sensory loss due to intra- and interfamilial phenotypic variability. This variation is seen even among families with the same genetic disorders.
Charcot-Marie-Tooth disease diagnosis
- Electrophysiology
Nerve conduction investigations aid in the confirmation of neuropathy diagnosis and the classification of patients into demyelinating and axonal subtypes. They are also beneficial in screening the index patient's asymptomatic relatives. Distal latencies, amplitudes, and velocities of motor and sensory nerves are the primary characteristics assessed. Conduction velocity slowing is an indirect marker of myelin impairment. Axonopathy is indicated by a reduction in the amplitude of the compound muscle action potential while maintaining the conduction velocity. Some individuals may have both demyelinating and axonal neuropathy symptoms. Concentric needle electromyography evidence of denervation is important in determining axonal disease.
- Nerve Imaging
Imaging advances have made it possible to see peripheral nerves clearly along their whole length. Nerve ultrasonography and magnetic resonance neurography are becoming more used in the diagnosis of neuropathies. There is widespread expansion in CMTs, encompassing roots, plexuses, and peripheral nerves, with no difference between entrapment and non-entrapment locations.
- Genetics
Genetic testing can help in establishing a definitive diagnosis (especially in sporadic patients), medical counseling, reproductive planning, and selecting patients for therapeutic trials and research. All patients should have an expert perform phenotypic characterization, extensive and precise pedigree analysis, and adequate pre-test counseling.
- Nerve Biopsy
In occasional situations, nerve biopsy can assist uncover the underlying genetic etiology, and it can help separate CMT from acquired illnesses like CIDP. When genetic testing finds "variants of unknown significance" or a new variant, nerve biopsy may also support a functional link. Morphological and ultrastructural abnormalities in axons, myelin, nodes of Ranvier, and mitochondria aid in understanding the activities of mutant genes and, to a lesser extent, the disease processes.
Charcot-Marie-Tooth disease Treatment
CMT therapy is largely rehabilitative and symptomatic because there is no definitive and effective disease-modifying treatment to alter the disease's inherent progressive course.
- Pharmacotherapy
The current understanding of the underlying genetic abnormality and pathophysiology of CMTs, combined with newer drug development techniques such as systemic biology-based modeling, anti-sense oligonucleotides, adenoviral vector-based drug delivery, and RNA interference technology, is the foundation of this strategy.
Other agents that have been studied for their potential therapeutic role in CMTs include those that promote axonal regeneration (neurotrophin-3, neuregulins), gene expression regulators (histone deacetylases), chaperones and inducers of heat shock proteins, calcium homeostasis neuroprotective and antioxidant drugs (purified polyols-resveratrol), and potassium channel blockers. Supplementation with essential fatty acids, phospholipids, vitamin E, creatine, and a combination of bovine-derived gangliosides was ineffective.
- Rehabilitation
It is the foundation of medical management and requires a multidisciplinary team. Patients are at risk of developing contractures and deformities, as well as a limited range of motion. Stretching exercises, aerobics, resistance training, and the timely use of orthotic devices are all key components of physical therapy. They increase and maintain muscular strength and function, as well as joint flexibility and range of motion, balance, and cardio-respiratory fitness.
There is also a beneficial effect on weariness and discomfort, as well as the prevention of stiffness and deformities. Patients have difficulties participating in the workout program in practice due to higher energy demand and changed gait kinematics. The idea of 'overuse weakness,' in which activity causes muscle loss and worsens weakness, is debatable in CMT.
In general, moderate or submaximal exercise is beneficial. An orthosis improves posture and walking balance in those who have ankle weakness or deformity. Hand muscular weakness need a supervised workout regimen as well as training for daily activities. Splinting helps to enhance hand dexterity.
Muscular dystrophy
The term "muscular dystrophy" refers to a group of genetic illnesses that cause progressive, widespread muscle disease caused by insufficient or absent glycoproteins in the muscle cell plasma membrane. Muscular dystrophy is a genetic condition with several variants. Each has its own inheritance pattern, onset duration, and rate of muscle loss.
Muscular dystrophy Causes
Muscular dystrophy is most commonly caused by faulty or missing glycoproteins in the muscular membrane. Each kind of muscular dystrophy is caused by a particular gene loss or mutation, resulting in a variety of enzymatic or metabolic abnormalities.
Types of muscular dystrophy
There are several forms of MD, each with somewhat distinct symptoms. Not all cause significant impairment, and many have no effect on life expectancy.
Some of the more common types of MD include:
- Duchenne MD – one of the most common and severe forms, it usually affects boys in early childhood; people with the condition will usually only live into their 20s or 30s
- Myotonic dystrophy - a kind of MD that can occur at any age; life expectancy isn't necessarily altered, although persons with severe myotonic dystrophy may live shorter lifetimes.
- Facioscapulohumeral MD - a kind of MD that can occur in childhood or age; it advances slowly and is seldom fatal.
- Becker MD - similar to Duchenne MD, but develops later in childhood and is less severe; life expectancy is typically not as impaired.
- Limb-girdle MD - a set of disorders that often appear in late childhood or early adulthood; some types grow swiftly and are life-threatening, while others progress slowly.
- Oculopharyngeal MD - a kind of MD that commonly develops between the ages of 50 and 60 and has no effect on life expectancy.
Symptoms of Duchenne muscular dystrophy
Duchenne muscular dystrophy is not usually noticeable before the age of two or three. Symptoms and signs include:
- Delayed walking age
- Frequent falls, difficulty rising up from the ground or going up hills or stairs
- Difficulty running and jumping
- Well-developed or excessively large calf muscles.
- A waddling walk
- A sway-back (‘lordosis’)
- A tendency to stand and walk on the forefoot, with the heel off the ground.
Diagnosing muscular dystrophy
There are several approaches for diagnosing the various forms of MD. The age at which the illness is identified varies depending on when the symptoms first occur.
Diagnosis will involve some or all of the following stages:
- Investigating any symptoms
- Discussing any family history of MD
- Physical examination
- Blood tests
- Electrical tests on the nerves and muscles
- A muscle biopsy (where a small tissue sample is removed for testing)
Consult your doctor if you or your kid exhibits any of the signs of MD. If required, they may refer you to a hospital for additional tests.
Treating muscular dystrophy
There's no cure for MD, but a range of treatments can help with the physical disabilities and problems that may develop. These can include:
- Mobility assistance – including exercise, physiotherapy and physical aids
- Support groups – to deal with the practical and emotional impact of MD
- Surgery – to correct postural deformities, such as scoliosis
- Medicine – such as steroids to improve muscle strength, or ACE inhibitors and beta blockers to treat heart problems
Spinal muscular atrophy (SMA)
Spinal muscular atrophy (SMA) is a hereditary illness that affects the central nervous system, peripheral nervous system, and voluntary muscle movement.
The spinal cord contains the majority of the nerve cells that regulate muscles, which is why the disease's name includes the term spinal. SMA is muscular in nature since it primarily affects muscles, which do not receive impulses from these nerve cells. Atrophy is the medical term meaning shrinking, which is what muscles do when they are not stimulated by nerve cells.
SMA is a motor neuron disease that includes the loss of nerve cells termed motor neurons in the spinal cord. The age at which SMA symptoms appear generally coincides with the severity of motor function impairment: The stronger the influence on motor function, the younger the age of onset. Typically, children who exhibit symptoms at birth or in infancy have the lowest degree of functioning.
What are the symptoms of SMA?
SMA symptoms vary in severity, ranging from moderate to severe. SMA generally affects the muscles closest to the center of the body (proximal muscles) more than the muscles further away from the center (distal muscles)
The predominant symptom of SMA is voluntary muscular weakness. The muscles closest to the center of the body, such as the shoulders, hips, thighs, and upper back, are the most impacted. The lower limbs appear to be more impacted than the upper limbs, and deep tendon reflexes are reduced.
If the muscles needed for breathing and swallowing are impaired, this results in anomalies in these functions. Spinal curvatures can develop if the back muscles weaken.
Amyotrophic lateral sclerosis
Amyotrophic lateral sclerosis (ALS), popularly known as "Lou Gehrig disease," is a motor neurological illness. Multiple paths (both heritable and sporadic) have been found to result in obviously identical disease entities, rather than a single cause.
ALS affects both upper and lower motor neurons and has a varied start pattern, most typically beginning with indications of lower motor neuron degeneration in the proximal limbs. Because it is a degenerative condition, it will certainly lead to paralysis and death. There is no cure for ALS; however, a combination of drugs and therapies can lessen symptoms and extend life, sometimes by 10 years or more.
Conclusion
Neuromuscular diseases are problems that are acquired or inherited (genetic) and affect some element of the neuromuscular system. A neuromuscular illness is defined as any condition that affects the peripheral nervous system (PNS), the neuromuscular junction, or skeletal muscle, which are all components of the motor unit. These are usually progressive in character, resulting in muscular weakening and weariness. These disorders are often manageable to enhance quality and duration of life, but they are incurable.